![]()
このサイトは、日本国内の医療関係者(医師、薬剤師、看護師、技師・技士等)を対象に、弊社が販売する医療用医薬品を適正にご使用いただくための情報を提供しています。国外の医療関係者、一般の方に対する情報提供を目的としたものではありませんのでご了承ください。
このサイトのご利用に際しては、協和キリンメディカルサイトのご利用条件が適用されます。

会員登録をしていない方
医療関係者の方は、製品基本情報と一部コンテンツをご覧いただけます。
医療機関にお勤めの皆様を対象とした「協和キリンメディカルサイト」では、詳しい製品関連情報、医療用麻薬情報、各領域の学術情報、お役立ちコンテンツなどをご利用いただけます。ぜひこの機会にお申し込みください。
新規会員登録
会員の方

2.禁忌(次の患者には投与しないこと)
「禁忌を含む注意事項等情報」は、電子添文をご参照ください。
本剤は一部承認外の成績を含む臨床試験成績に基づき承認されました。
よって紹介する臨床成績には一部承認外の効能又は効果、用法及び用量の成績が含まれます。
国内第Ⅲ相臨床試験
有効性協和キリン株式会社:承認時評価資料 第Ⅲ相二重盲検試験(国内・パーキンソン病患者)
観察期の1日平均オフ時間はプラセボ群で6.31時間、ノウリアスト®20mg 群で6.55時間、40mg 群で5.97時間でした。
最終評価時の観察期からの変化(最小二乗平均値[95%C.I.])は、プラセボ群−0.23時間(−0.62、0.16)、ノウリアスト®20mg 群−0.99時間(−1.38、−0.60)、40mg 群−0.96時間(−1.35、−0.58)であり、プラセボ群に対するノウリアスト®20mg群、40mg群の優越性が検証されました(検証的解析結果、p=0.003、Williams検定、有意水準片側2.5%)。
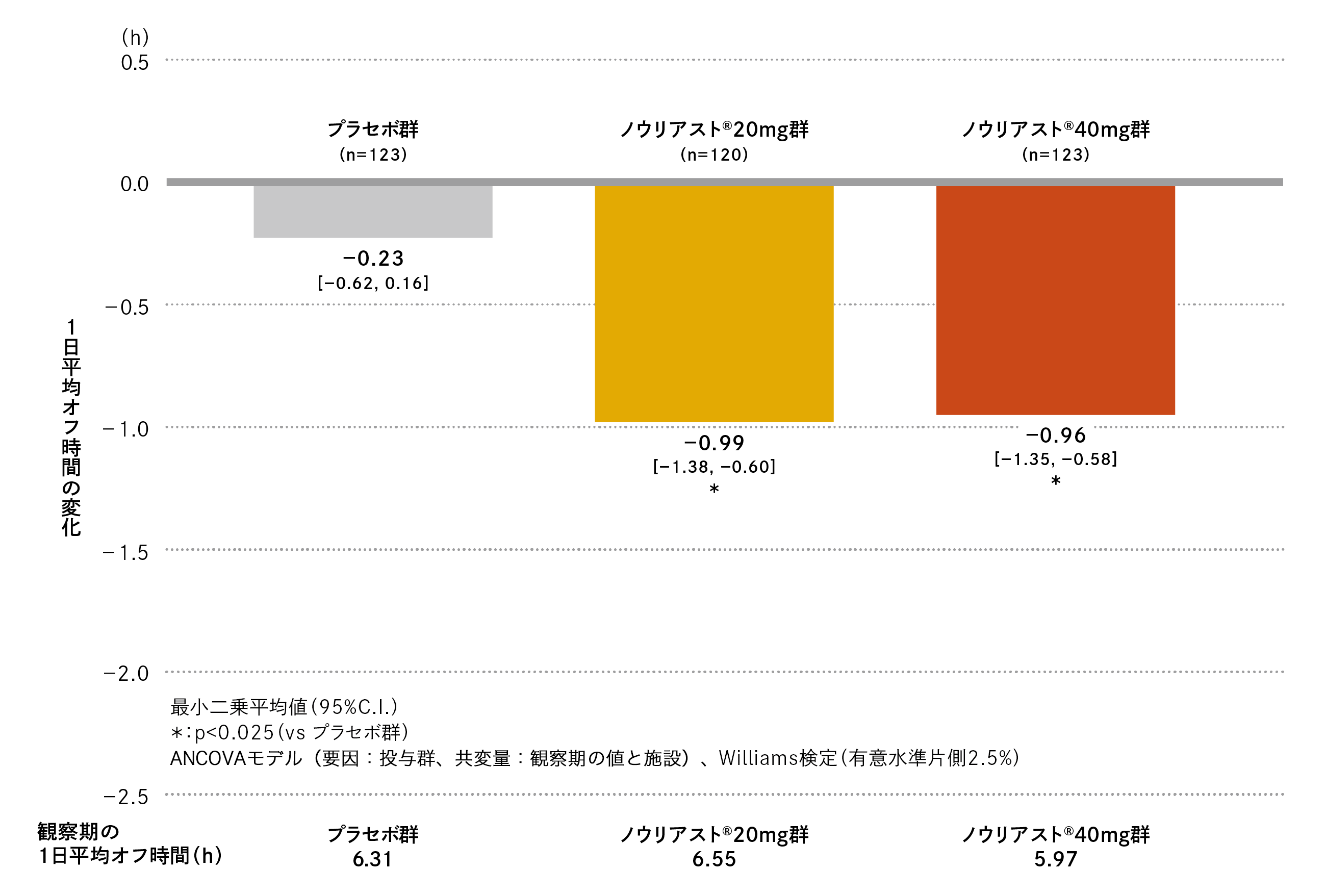
1日平均オフ時間の変化の推移は以下の通りでした。
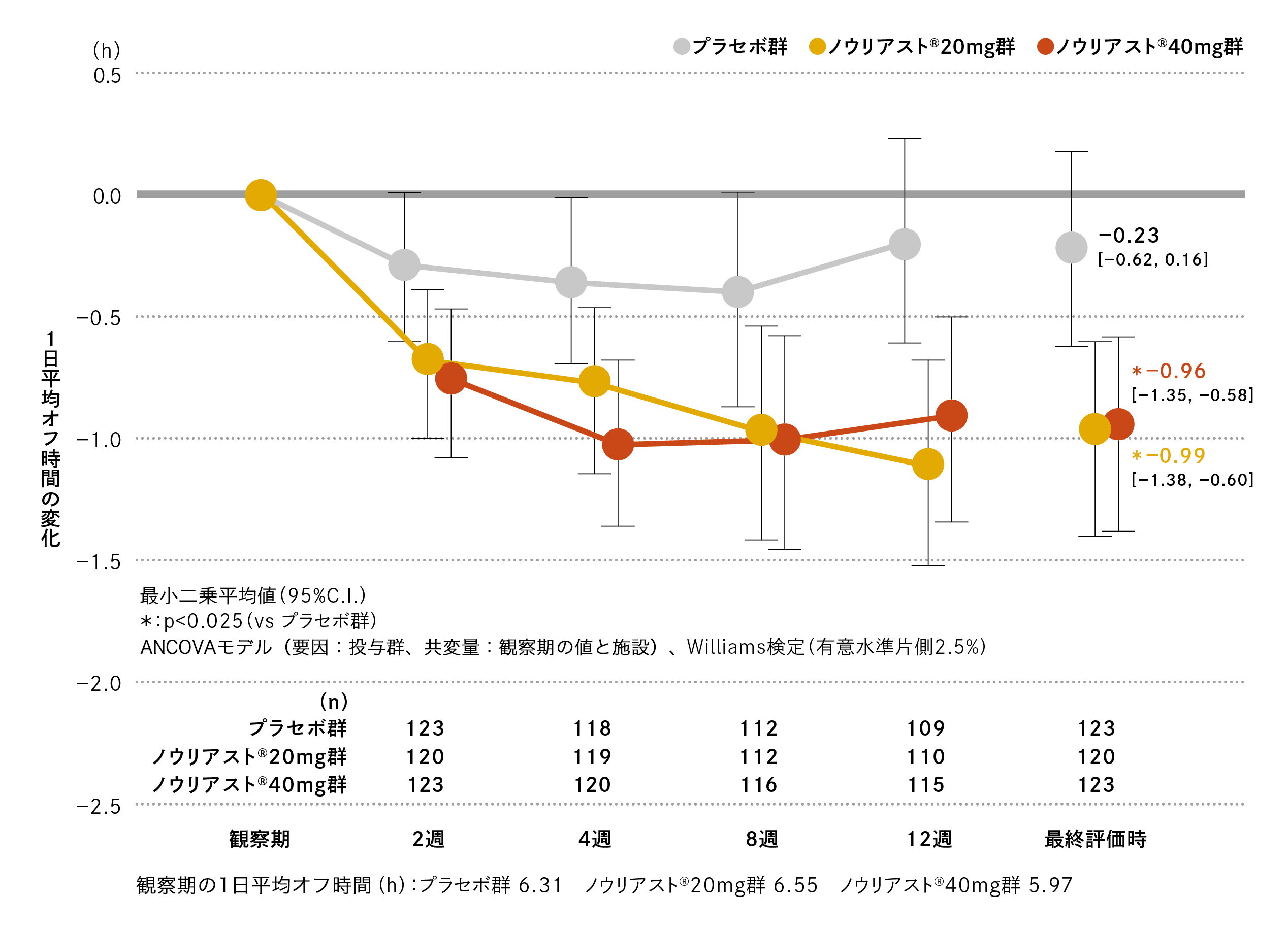
観察期の1日平均オフ時間はプラセボ群で6.31時間、ノウリアスト®20mg 群で6.55時間、40mg 群で5.97時間でした。
最終評価時の観察期からの変化(最小二乗平均値[95%C.I.])は、プラセボ群−0.23時間(−0.62、0.16)、ノウリアスト®20mg 群−0.99時間(−1.38、−0.60)、40mg 群−0.96時間(−1.35、−0.58)であり、プラセボ群に対するノウリアスト®20mg群、40mg群の優越性が検証されました(検証的解析結果、p=0.003、Williams検定、有意水準片側2.5%)。
国内第Ⅲ相臨床試験
有効性協和キリン株式会社:承認時評価資料 第Ⅲ相二重盲検試験(国内・パーキンソン病患者)
観察期におけるオフ時のUPDRS part Ⅱスコアは、プラセボ群、ノウリアスト®20mg 群、40mg 群でそれぞれ14.7、14.9、15.4でした。最終評価時の観察期からの変化(最小二乗平均値[95%C.I.])は、プラセボ群−0.6(−1.2、−0.1)、ノウリアスト®20mg 群−1.4(−2.0、−0.8)、40mg 群−1.7(−2.2、−1.1)であり、プラセボ群に対してノウリアスト®40mg 群では有意差が認められました(名目上のp値=0.009、Williams検定、有意水準片側2.5%)。
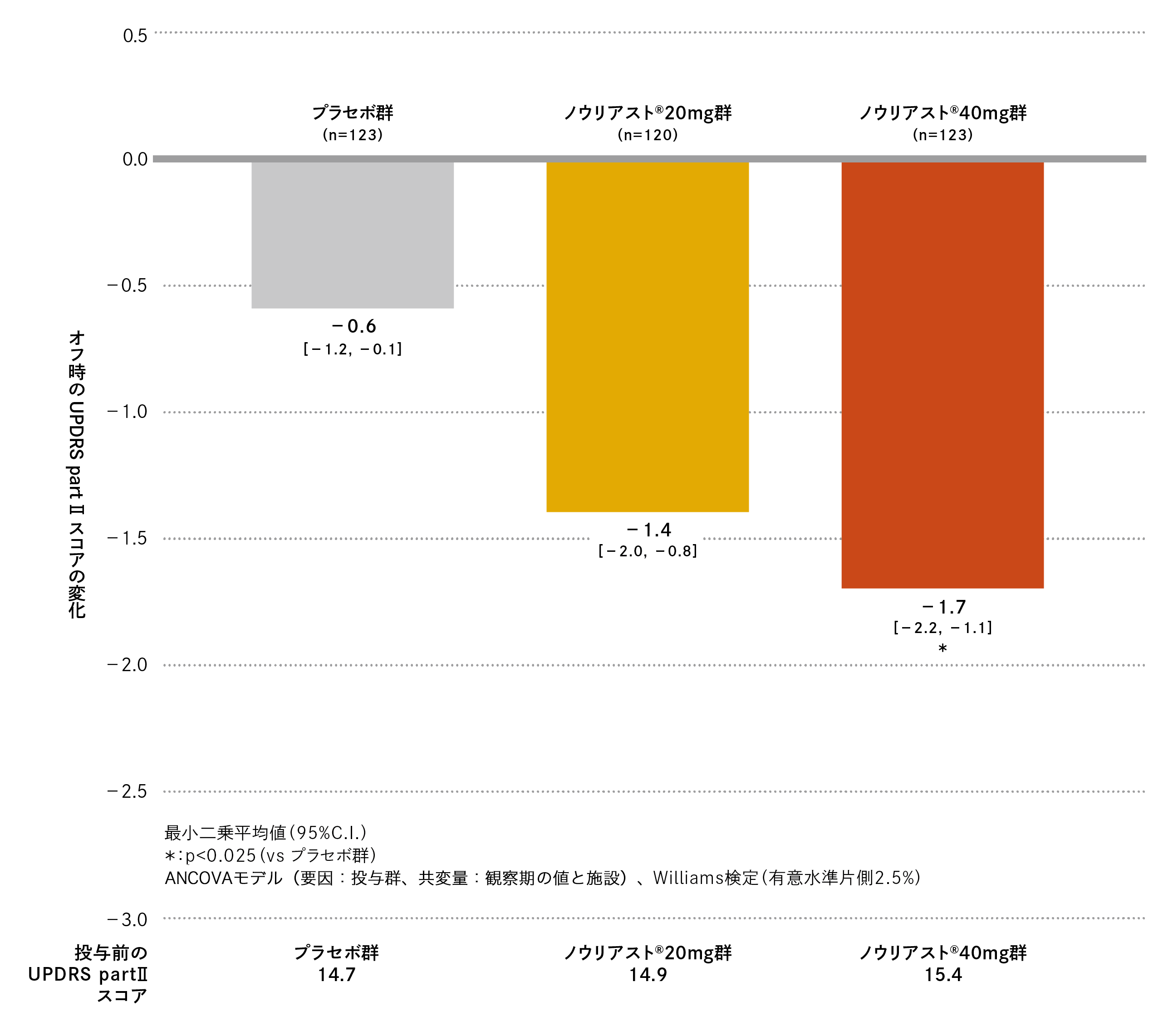
オフ時のUPDRS part Ⅱスコアの変化の推移は以下の通りでした。
観察期におけるオフ時のUPDRS part Ⅱスコアは、プラセボ群、ノウリアスト®20mg 群、40mg 群でそれぞれ14.7、14.9、15.4でした。最終評価時の観察期からの変化(最小二乗平均値[95%C.I.])は、プラセボ群−0.6(−1.2、−0.1)、ノウリアスト®20mg 群−1.4(−2.0、−0.8)、40mg 群−1.7(−2.2、−1.1)であり、プラセボ群に対してノウリアスト®40mg 群では有意差が認められました(名目上のp値=0.009、Williams検定、有意水準片側2.5%)。
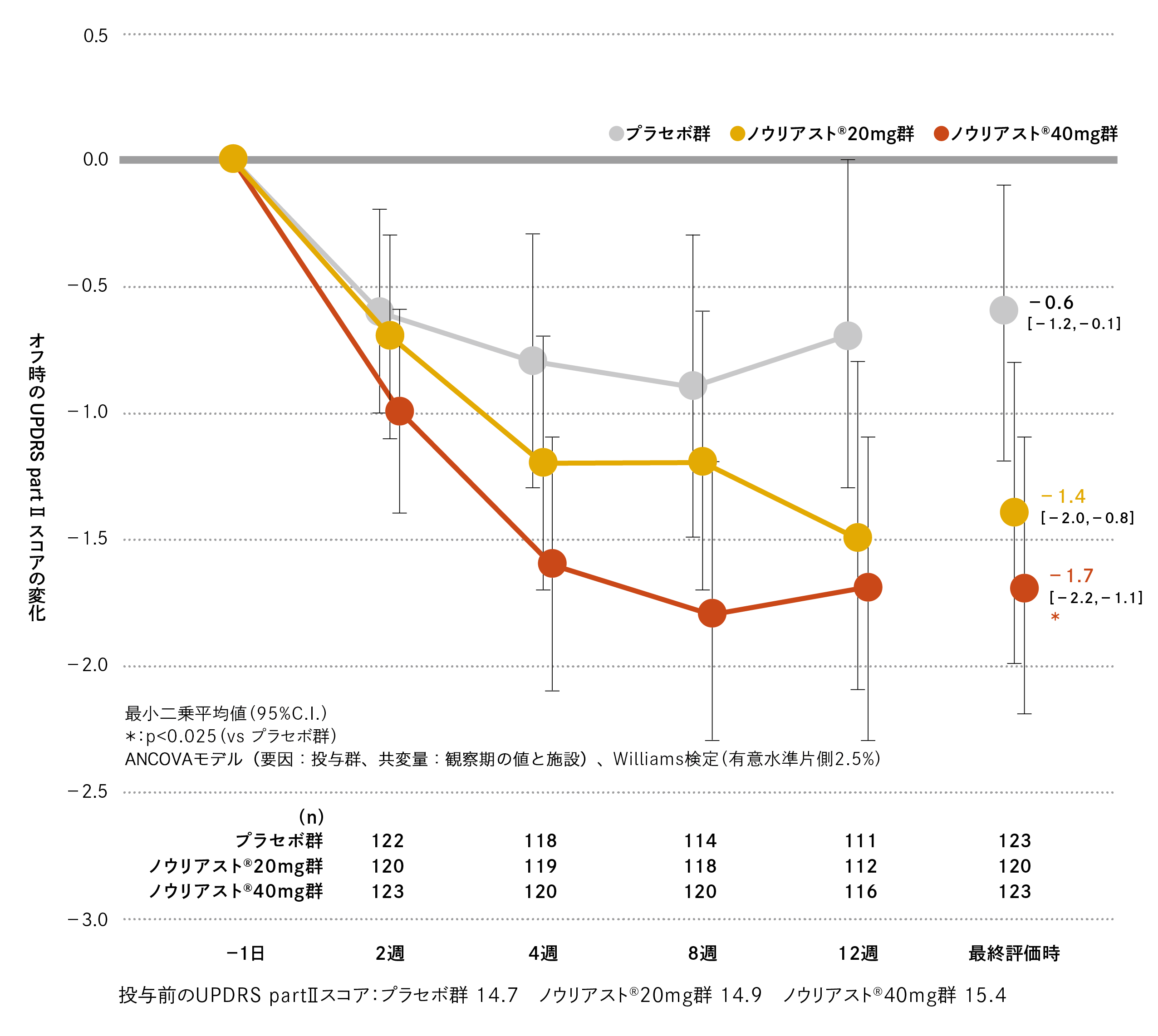
国内第Ⅲ相臨床試験
有効性協和キリン株式会社:承認時評価資料 第Ⅲ相二重盲検試験(国内・パーキンソン病患者)
観察期におけるオン時のUPDRS part Ⅲスコアは、プラセボ群、ノウリアスト®20mg 群、40mg 群でそれぞれ21.6、21.3、20.7でした。最終評価時の観察期からの変化(最小二乗平均値[95%C.I.])は、プラセボ群−2.8(−3.8、−1.8)、ノウリアスト®20mg 群−3.7(−4.7、−2.8)、40mg 群−4.9(−5.8、−3.9)であり、ノウリアスト®40mg群はプラセボ群に比べてオン時のUPDRS part Ⅲスコアにて有意差が認められました(名目上のp値=0.001、Williams検定、有意水準片側2.5%)。
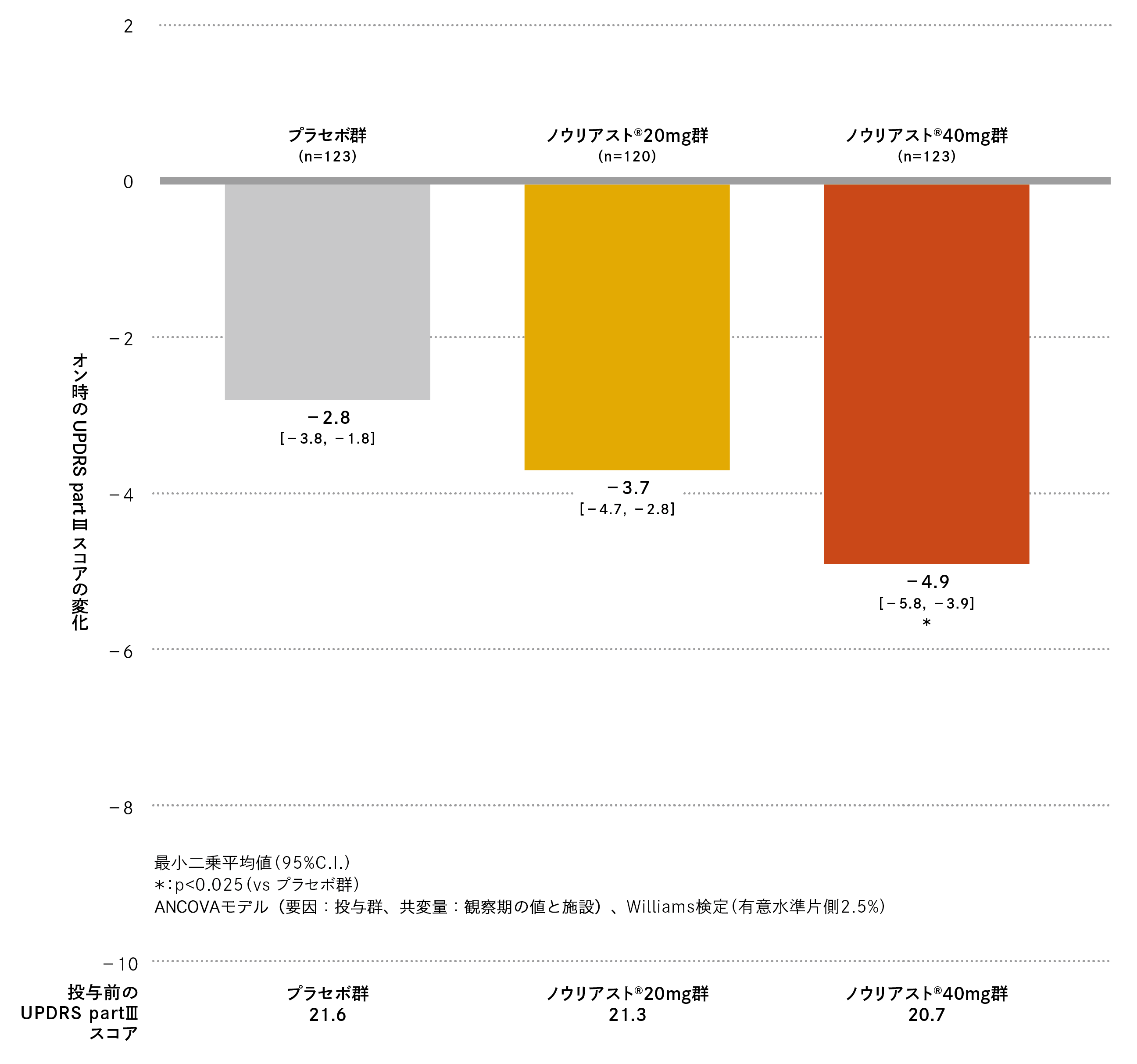
オン時のUPDRS part Ⅲスコアの変化の推移は以下の通りでした。
観察期におけるオン時のUPDRS part Ⅲスコアは、プラセボ群、ノウリアスト®20mg 群、40mg 群でそれぞれ21.6、21.3、20.7でした。最終評価時の観察期からの変化(最小二乗平均値[95%C.I.])は、プラセボ群−2.8(−3.8、−1.8)、ノウリアスト®20mg 群−3.7(−4.7、−2.8)、40mg 群−4.9(−5.8、−3.9)であり、ノウリアスト®40mg群はプラセボ群に比べてオン時のUPDRS part Ⅲスコアにて有意差が認められました(名目上のp値=0.001、Williams検定、有意水準片側2.5%)。
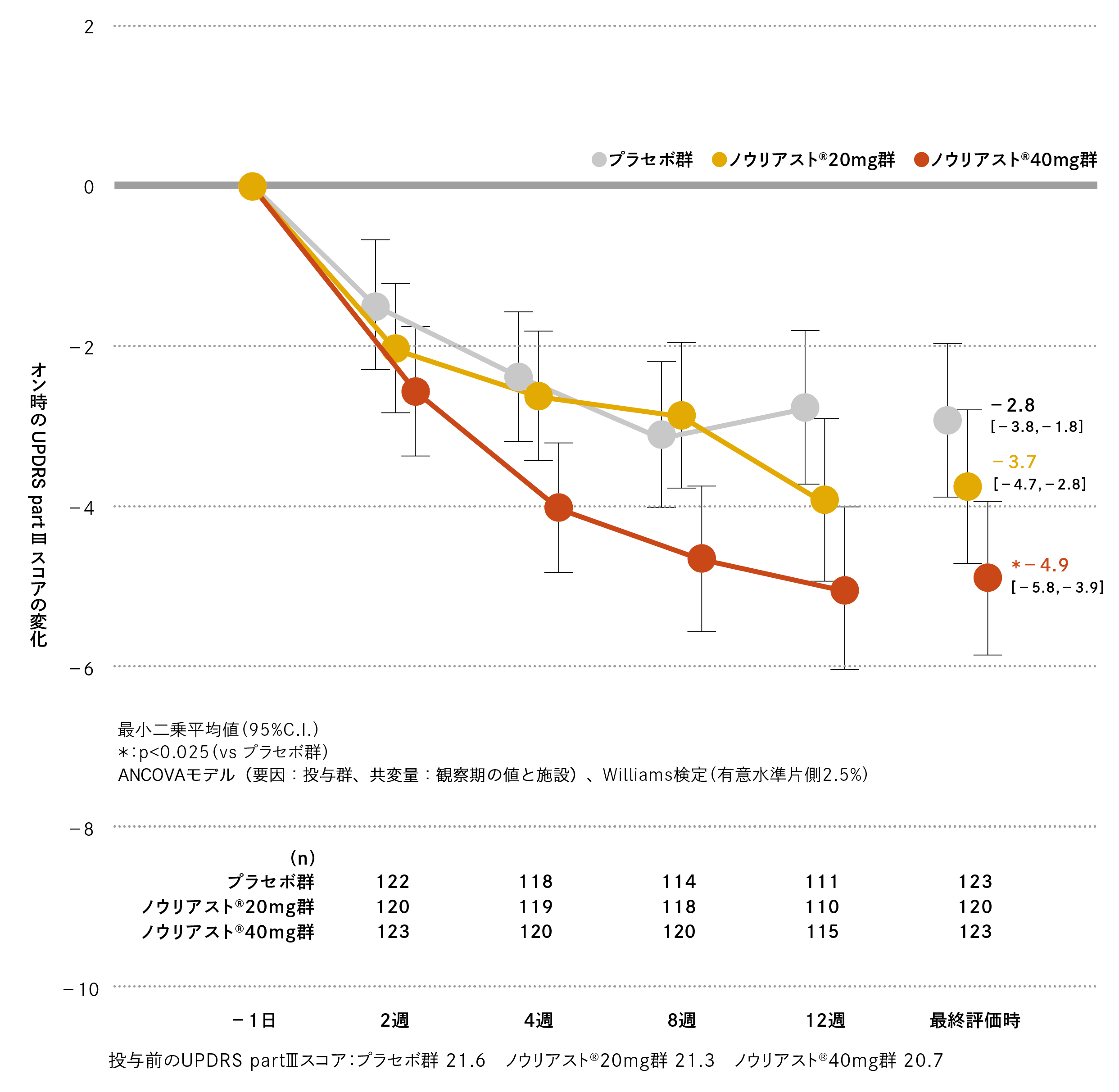
国内第Ⅲ相臨床試験
安全性協和キリン株式会社:承認時評価資料 第Ⅲ相二重盲検試験(国内・パーキンソン病患者)
本試験における副作用発現率はプラセボ群28.6%(36/126例)、ノウリアスト®20mg 群36.6%(45/123例)、ノウリアスト®40mg 群38.7%(48/124例)でした。主な副作用はジスキネジーでそれぞれ4.0%(5/126例)、12.2%(15/123例)、12.1%(15/124例)でした。重篤な副作用はノウリアスト®20mg 群で歩行障害及びパーキンソニズムがそれぞれ1例、ノウリアスト®40mg群で胃潰瘍、心筋梗塞及び幻覚がそれぞれ1例でした。投与中止に至った副作用は、プラセボ群では、丘疹性皮疹、幻覚、体幹前屈症、パーキンソン歩行、流涎過多、無力症が各1例、ノウリアスト®20mg 群では、過眠症、起立性低血圧、胸部不快感、浮動性めまいが各1例、ノウリアスト®40mg 群ではジスキネジーが2例、幻視、激越、体感幻覚、躁病、不安が各1例でした。本試験において、プラセボ群で1例に死亡(原因不明)が報告されました。本試験においてノウリアスト®20mg 群、40mg 群では死亡に至った副作用は認められませんでした。
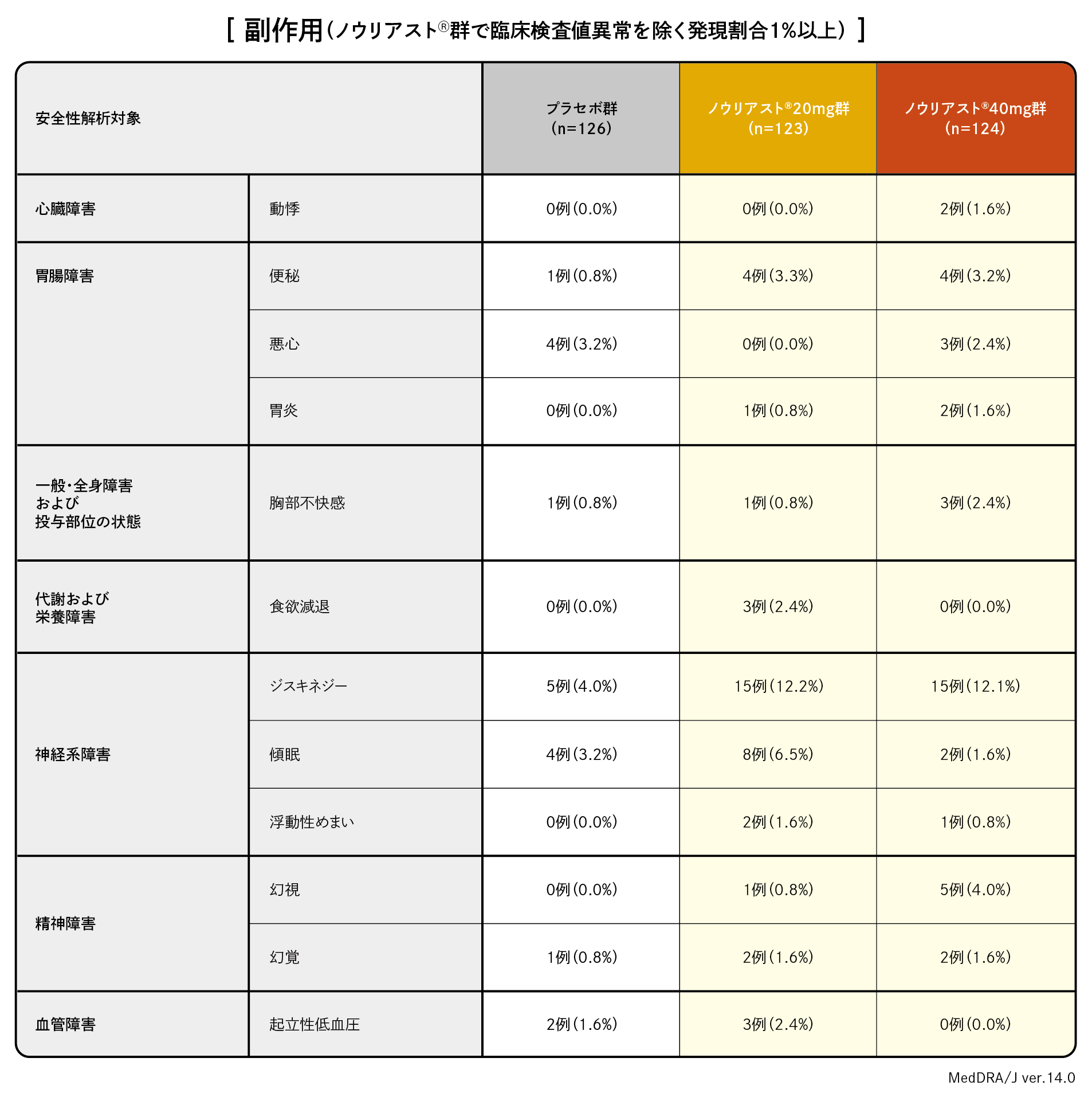
| 【目的】 | パーキンソン病患者に対し、ノウリアスト®を1日1回投与した際の有効性をプラセボと比較し検証する。また、安全性についても検討する。 |
|---|---|
| 【対象】 | レボドパ含有製剤で治療中の運動合併症(ウェアリングオフ現象)を併発しているパーキンソン病患者373例
|
| 【方法】 | 第Ⅲ相臨床試験(多施設共同プラセボ対照無作為化二重盲検並行群間比較試験) 対象患者を無作為割り付けし、既存の経口薬に加え、プラセボ、ノウリアスト®20mg、40mgを1日1回12週間経口投与した。 |
| 【観察期】 | 無作為化時より前の2〜4週間 |
| 【主要評価項目】 |
|
| 【副次評価項目】 |
|
| 【安全性評価項目】 | 有害事象、副作用の有無及び内容 |
| 【解析計画】 | ●主要評価項目 1日平均オフ時間に対する主たる解析はANCOVAモデルによるものとした。投与後の各評価時点又は最終評価時と観察期における1日平均オフ時間の差に対して、投与群を要因、観察期の1日平均オフ時間と施設を共変量としたANCOVAモデルをあてはめた。ANCOVAモデルのもとで、1日平均オフ時間の差の推定値として、投与群ごとに最小二乗平均値と95%信頼区間(95%C.I.)を算出した。ノウリアスト®20mg群及び40mg群それぞれについて、プラセボ群に対する差の最小二乗平均値と95%C.I.を算出した。 最終評価時において、プラセボ群とノウリアスト®20mg群及び40mg群の比較をWilliams検定により有意水準片2.5%で実施し、用量反応関係を想定してプラセボ群に対するノウリアスト®群の効果を検証した(検証的解析)。 ●副次評価項目 状態別の1日平均オン時間及びオフ時のUPDRS part Ⅱスコア、オン時のUPDRS part Ⅲスコアについては、1日平均オフ時間に対する解析と同様に、観察期の値との差を応答変数、投与群を要因、観察期の値と施設を共変量としたANCOVAモデルをあてはめ、投与群ごとに最小二乗平均値と95%C.I.を算出した。また、ノウリアスト®20mg群及び40mg群それぞれについて、プラセボ群に対する差の最小二乗平均値と95%C.I.を算出した。 ●安全性 治験薬投与後に発現したすべての有害事象を対象として、有害事象、因果関係が否定できない有害事象の有無及び内容別の発現頻度を投与群別に集計した。 |
| 【判断基準】 | ●1日覚醒時間における平均オフ時間 症状日誌において「オフ」にチェックされたマスの数×0.5時間として評価した。 |
6. 用法及び用量
7. 用法及び用量に関連する注意
7.1
患者のオン時の運動機能の改善を期待する場合、40mgを1日1回経口投与できる。ただし、40mgでは、20mgを上回るオフ時間の短縮効果は認められていない。[17.1.1参照]
7.2
以下の患者では本剤の血中濃度が上昇するおそれがあるため、1日1回20mgを上限とすること。
9. 特定の背景を有する患者に関する注意(抜粋)
9.1
合併症・既往歴等のある患者
9.1.2
ジスキネジーのある患者
患者の状態を注意深く観察しながら投与すること。ジスキネジーを悪化させることがある。ジスキネジーが悪化した場合には必要に応じ、本剤の減量、休薬又は投与中止等の適切な処置を行うこと。
本剤は一部承認外の成績を含む臨床試験成績に基づき承認されました。
よって紹介する臨床成績には一部承認外の効能又は効果、用法及び用量の成績が含まれます。
プラセボ対照比較試験 3試験の併合解析
ノウリアスト®の安全性承認時評価資料 : プラセボ対照比較試験3試験(前期第Ⅱ相、後期第Ⅱ相、第Ⅲ相)の併合解析(国内・パーキンソン病患者)
プラセボ対照比較試験(3試験の併合解析)において、臨床検査値異常を含む副作用は、プラセボ群で275例中89例(32.4%)、ノウリアスト®20mg群で272例中104例(38.2%)、40mg群で277例中107例(38.6%)に認められました。主な副作用はジスキネジーでそれぞれ3.3%、11.0%、10.1%でした。
重篤な副作用はノウリアスト®20mg群で胃潰瘍及び歩行障害、パーキンソニズムがそれぞれ1例、ノウリアスト®40mg群で幻覚及び被害妄想がそれぞれ2例、心筋梗塞及び胃潰瘍、うつ病、咳嗽、高血圧がそれぞれ1例でした。プラセボ群では一過性脳虚血発作が1例でした。
投与中止に至った副作用は、前期第Ⅱ相試験のノウリアスト®20mg群(n=31)でジスキネジーが1例、ノウリアスト®40mg群(n=28)でジスキネジーが2例、悪心、味覚異常、食欲減退、体重減少、パーキンソン病、頭痛、発熱、咽頭紅斑がそれぞれ1例、後期第Ⅱ相試験のプラセボ群(n=119)で異常感が1例、ノウリアスト®20mg群(n=118)でジスキネジーが3例、蕁麻疹、パーキンソン病、不眠症、四肢痛がそれぞれ1例、ノウリアスト®40mg 群(n=125)で幻覚、アスパラギン酸アミノトランスフェラーゼ増加、血中アルカリホスファターゼ増加がそれぞれ2例、心房細動、ジスキネジー、食欲減退、アレルギー性皮膚炎、ブドウ膜炎、γ-グルタミルトランスフェラーゼ増加、アラニン・アミノトランスフェラーゼ増加、血中クレアチンホスホキナーゼ増加、血中乳酸脱水素酵素増加、妄想がそれぞれ1例、第Ⅲ相試験のプラセボ群(n=126)で丘疹性皮疹、幻覚、体幹前屈症、パーキンソン歩行、流涎過多、無力症がそれぞれ1例、ノウリアスト®20mg 群(n=123)で過眠症、起立性低血圧、胸部不快感、浮動性めまいがそれぞれ1例、ノウリアスト®40mg 群(n=124)でジスキネジーが2例、幻視、激越、体感幻覚、躁病、不安がそれぞれ1例でした。また、第Ⅲ相試験のプラセボ群で1例に死亡が報告されました。
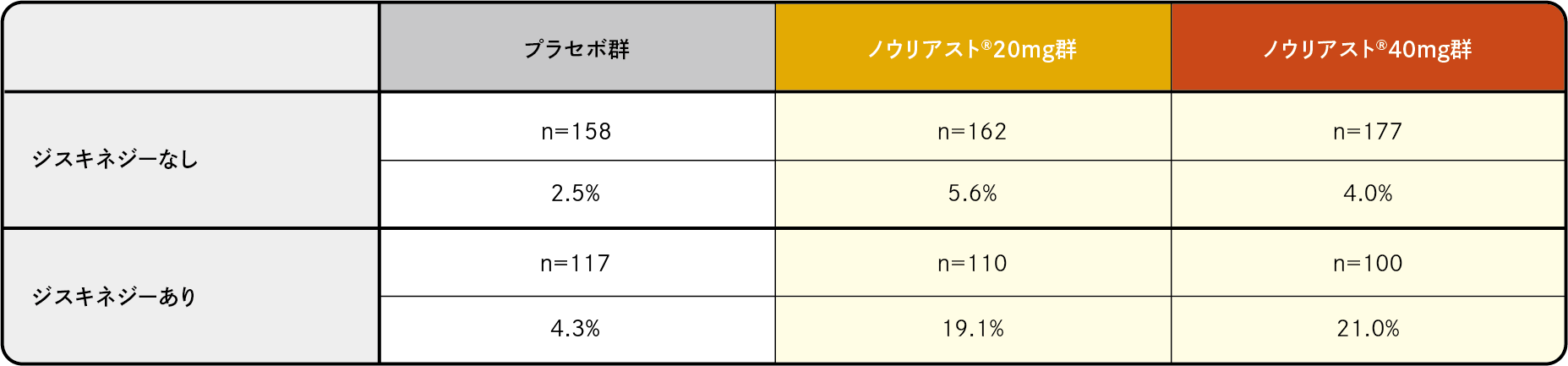
プラセボ対照比較試験(3試験の併合解析)において、臨床検査値異常を含む副作用は、65歳未満ではノウリアスト®20mg群、40mg群それぞれ、116例中48例(41.4%)、125例中46例(36.8%)、プラセボ群129例中43例(33.3%)、65歳以上ではノウリアスト®20mg群、40mg群それぞれ、156例中56例(35.9%)、152例中61例(40.1%)、プラセボ群146例中46例(31.5%)に認められました。
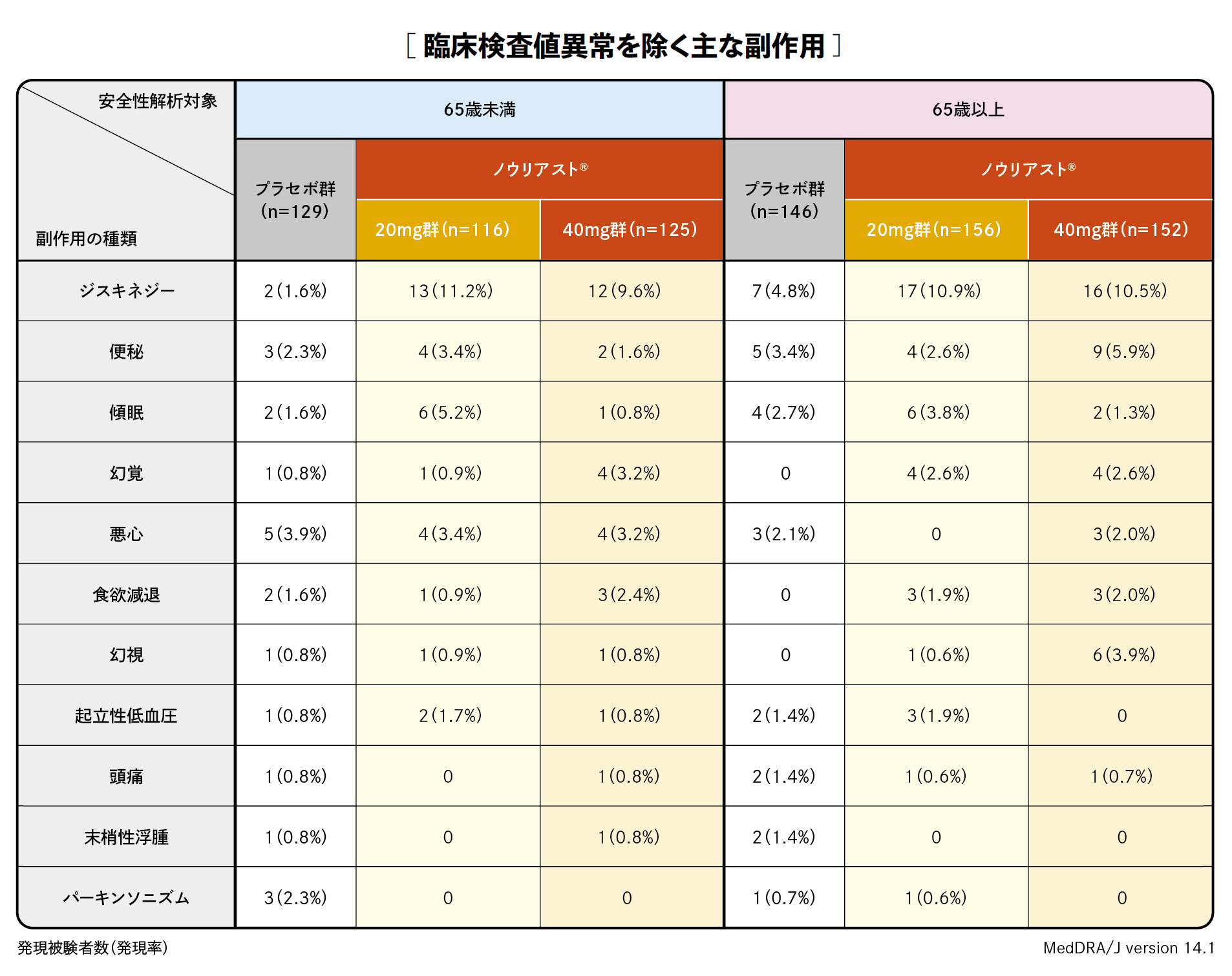
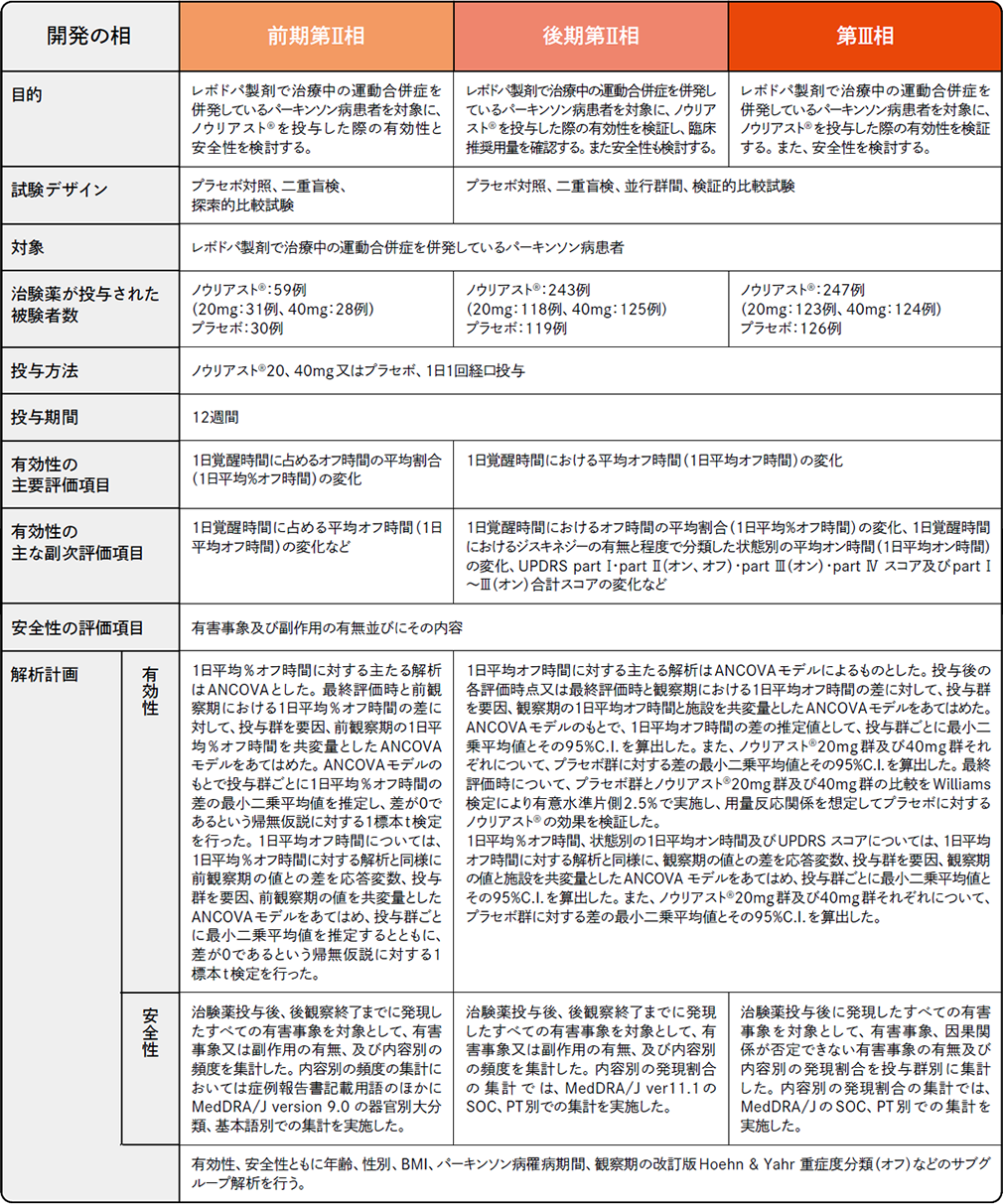
プラセボ対照比較国内臨床試験
各試験の試験デザイン承認時評価資料 : プラセボ対照比較試験(前期第Ⅱ相、後期第Ⅱ相、第Ⅲ相)(国内・パーキンソン病患者)
プラセボ対照比較国内臨床試験
各試験の患者背景(FAS)承認時評価資料 : プラセボ対照比較試験(前期第Ⅱ相、後期第Ⅱ相、第Ⅲ相)(国内・パーキンソン病患者)
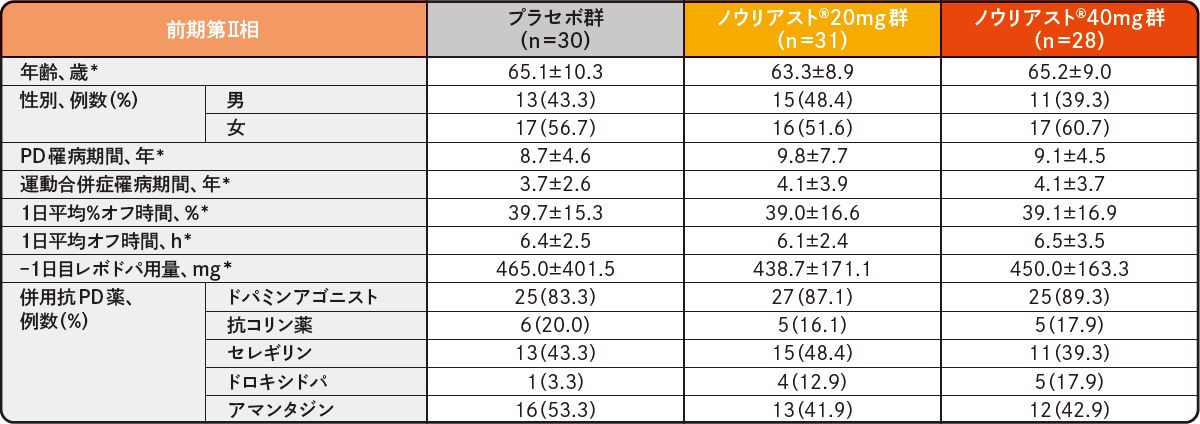
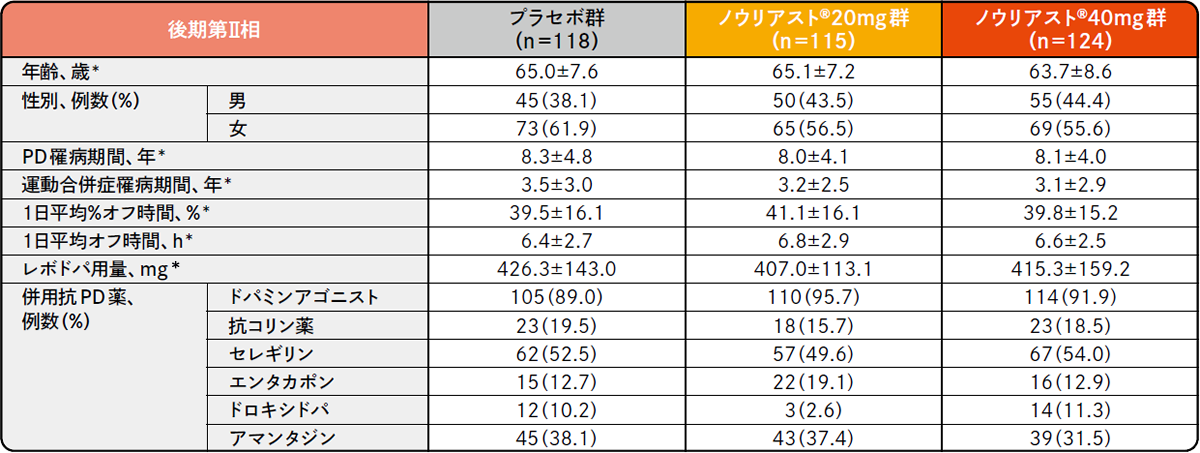
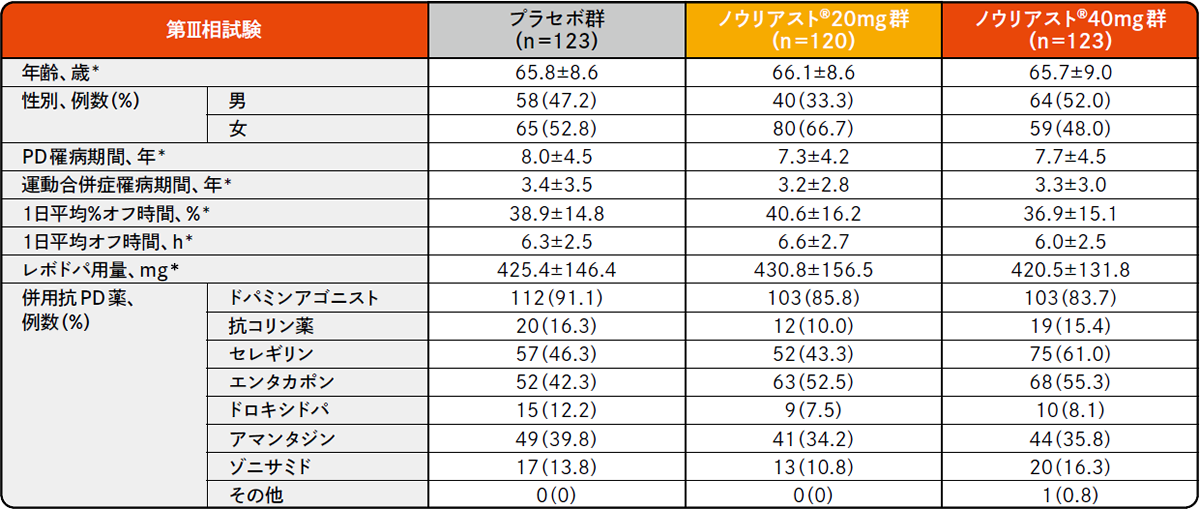
*平均値±SD
| 【目的】 | パーキンソン病患者に対しノウリアスト®を投与した際、患者背景が有効性及び安全性に及ぼす影響を検討する。 |
|---|---|
| 【試験デザイン】 | 二重盲検、プラセボ対照比較試験 (治験実施計画に基づいて実施された3試験の併合解析) |
| 【解析対象集団】 | 安全性(各試験のSS) |
| 【観察期間】 | 12週 |
| 【評価項目】 | 安全性など |
| 【解析計画】 | 安全性は、3つのプラセボ対照比較試験でノウリアスト®を1回以上投与した患者を対象に解析した。サブグループ解析として、年齢別にみた副作用(事前規程)、ジスキネジー有無別のジスキネジー発症率(審査の過程で照会事項に対する回答として提出され、了承された解析)を集計した。 |
6. 用法及び用量
7. 用法及び用量に関連する注意
7.1
患者のオン時の運動機能の改善を期待する場合、40mgを1日1回経口投与できる。ただし、40mgでは、20mgを上回るオフ時間の短縮効果は認められていない。[17.1.1参照]
7.2
以下の患者では本剤の血中濃度が上昇するおそれがあるため、1日1回20mgを上限とすること。
9. 特定の背景を有する患者に関する注意(抜粋)
9.1
合併症・既往歴等のある患者
9.1.2
ジスキネジーのある患者
患者の状態を注意深く観察しながら投与すること。ジスキネジーを悪化させることがある。ジスキネジーが悪化した場合には必要に応じ、本剤の減量、休薬又は投与中止等の適切な処置を行うこと。
10. 相互作用
10.2
併用注意(併用に注意すること)(抜粋)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
| エンタカポン | エンタカポンとの併用によりジスキネジーの発現頻度の上昇が認められた。 | 機序は不明である。 |
2024年11月作成
KKC-2021-00466-4
Copyright © Kyowa Kirin Co., Ltd. All rights reserved.